AlphaFold


In July 2022, we released AlphaFold protein structure predictions for nearly all catalogued proteins known to science. Read the latest blog here.
Proteins are essential to life, supporting practically all its functions. They are large complex molecules, made up of chains of amino acids, and what a protein does largely depends on its unique 3D structure. Figuring out what shapes proteins fold into is known as the “protein folding problem”, and has stood as a grand challenge in biology for the past 50 years. In a major scientific advance, the latest version of our AI system AlphaFold has been recognised as a solution to this grand challenge by the organisers of the biennial Critical Assessment of protein Structure Prediction (CASP). This breakthrough demonstrates the impact AI can have on scientific discovery and its potential to dramatically accelerate progress in some of the most fundamental fields that explain and shape our world.
A protein’s shape is closely linked with its function, and the ability to predict this structure unlocks a greater understanding of what it does and how it works. Many of the world’s greatest challenges, like developing treatments for diseases or finding enzymes that break down industrial waste, are fundamentally tied to proteins and the role they play.
We have been stuck on this one problem – how do proteins fold up – for nearly 50 years. To see DeepMind produce a solution for this, having worked personally on this problem for so long and after so many stops and starts, wondering if we’d ever get there, is a very special moment.
This has been a focus of intensive scientific research for many years, using a variety of experimental techniques to examine and determine protein structures, such as nuclear magnetic resonance and X-ray crystallography. These techniques, as well as newer methods like cryo-electron microscopy, depend on extensive trial and error, which can take years of painstaking and laborious work per structure, and require the use of multi-million dollar specialised equipment.
In his acceptance speech for the 1972 Nobel Prize in Chemistry, Christian Anfinsen famously postulated that, in theory, a protein’s amino acid sequence should fully determine its structure. This hypothesis sparked a five decade quest to be able to computationally predict a protein’s 3D structure based solely on its 1D amino acid sequence as a complementary alternative to these expensive and time consuming experimental methods. A major challenge, however, is that the number of ways a protein could theoretically fold before settling into its final 3D structure is astronomical. In 1969 Cyrus Levinthal noted that it would take longer than the age of the known universe to enumerate all possible configurations of a typical protein by brute force calculation – Levinthal estimated 10^300 possible conformations for a typical protein. Yet in nature, proteins fold spontaneously, some within milliseconds – a dichotomy sometimes referred to as Levinthal’s paradox.
In 1994, Professor John Moult and Professor Krzysztof Fidelis founded CASP as a biennial blind assessment to catalyse research, monitor progress, and establish the state of the art in protein structure prediction. It is both the gold standard for assessing predictive techniques and a unique global community built on shared endeavour. Crucially, CASP chooses protein structures that have only very recently been experimentally determined (some were still awaiting determination at the time of the assessment) to be targets for teams to test their structure prediction methods against; they are not published in advance. Participants must blindly predict the structure of the proteins, and these predictions are subsequently compared to the ground truth experimental data when they become available. We’re indebted to CASP’s organisers and the whole community, not least the experimentalists whose structures enable this kind of rigorous assessment.
The main metric used by CASP to measure the accuracy of predictions is the Global Distance Test (GDT) which ranges from 0-100. In simple terms, GDT can be approximately thought of as the percentage of amino acid residues (beads in the protein chain) within a threshold distance from the correct position. According to Professor Moult, a score of around 90 GDT is informally considered to be competitive with results obtained from experimental methods.
In the results from the 14th CASP assessment, released today, our latest AlphaFold system achieves a median score of 92.4 GDT overall across all targets. This means that our predictions have an average error (RMSD) of approximately 1.6 Angstroms, which is comparable to the width of an atom (or 0.1 of a nanometer). Even for the very hardest protein targets, those in the most challenging free-modelling category, AlphaFold achieves a median score of 87.0 GDT (data available here).

Improvements in the median accuracy of predictions in the free modelling category for the best team in each CASP, measured as best-of-5 GDT.
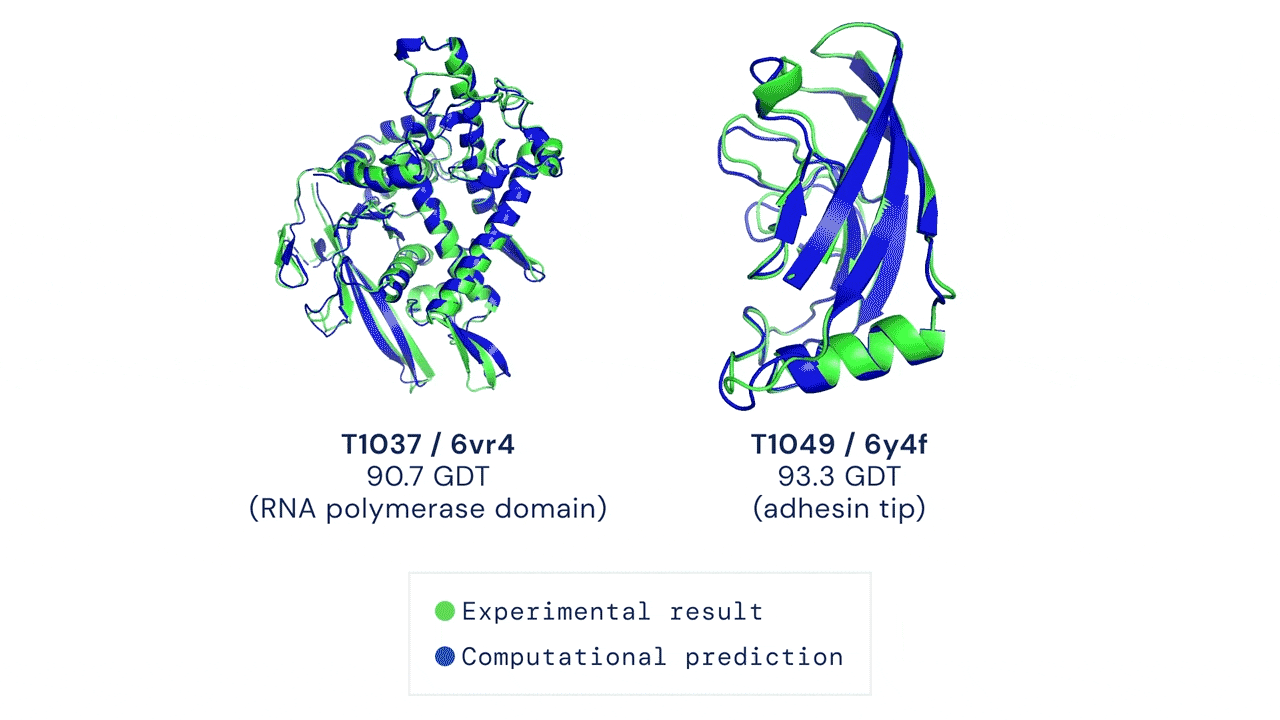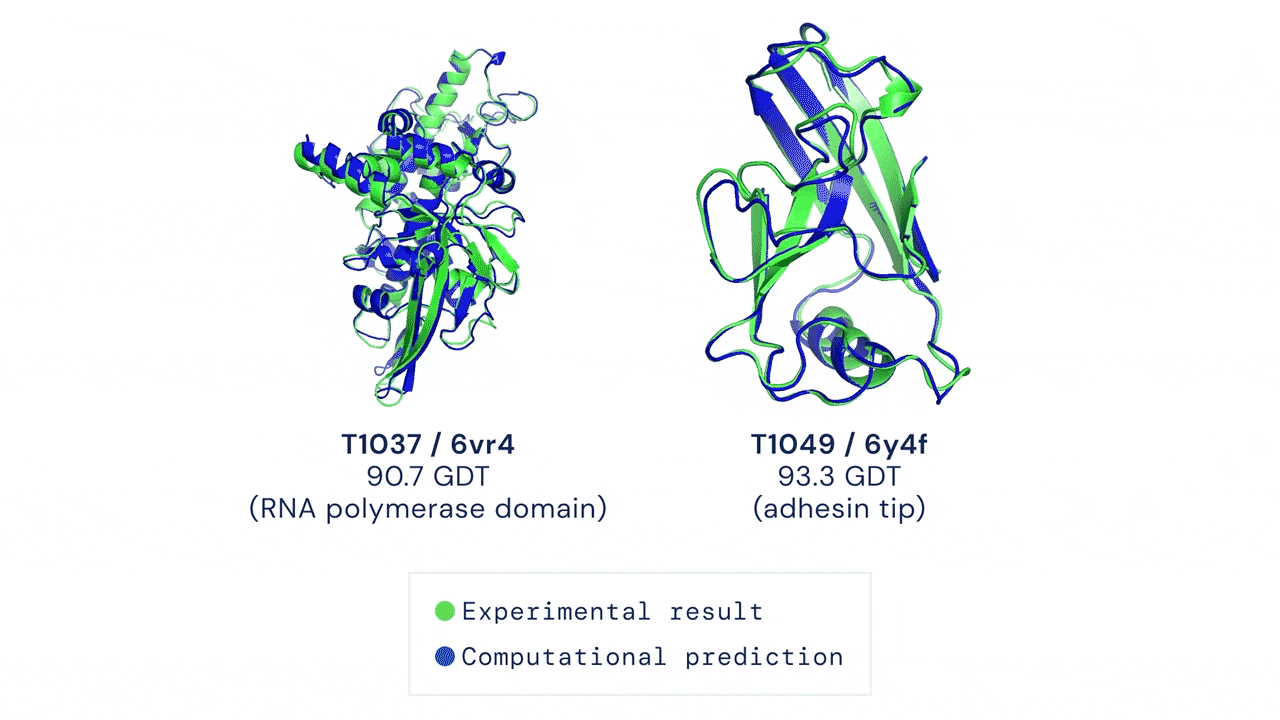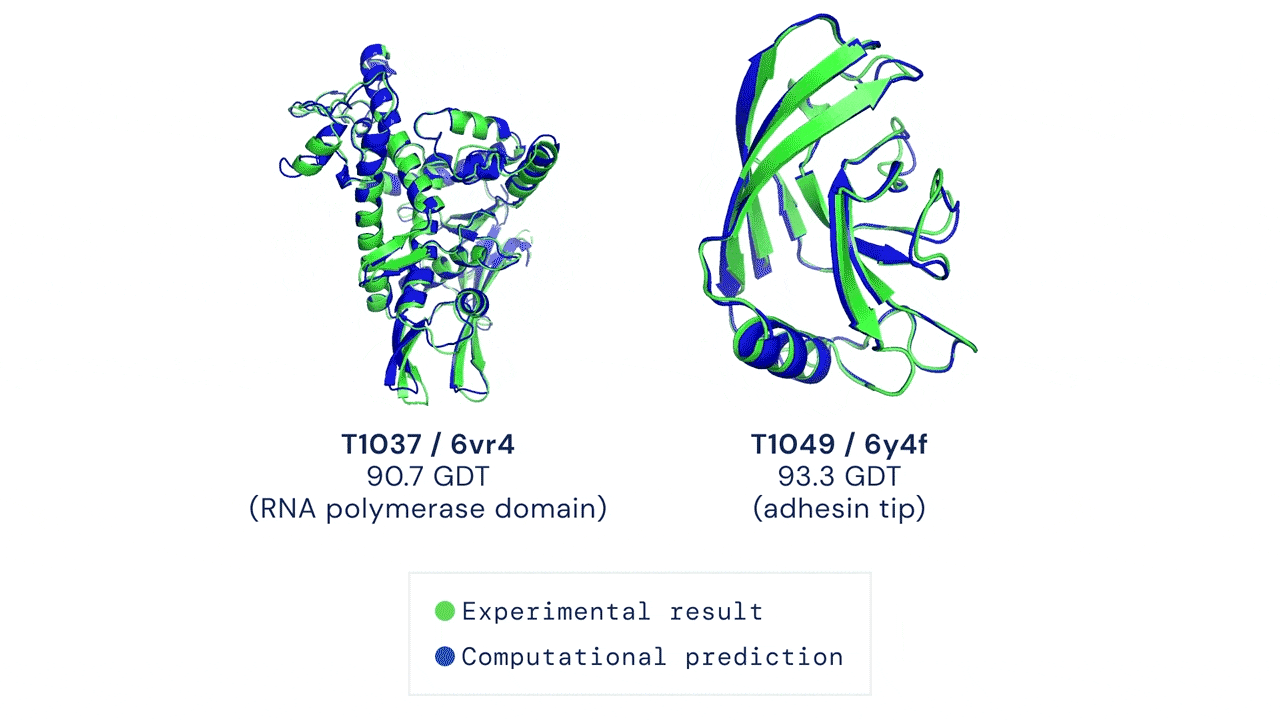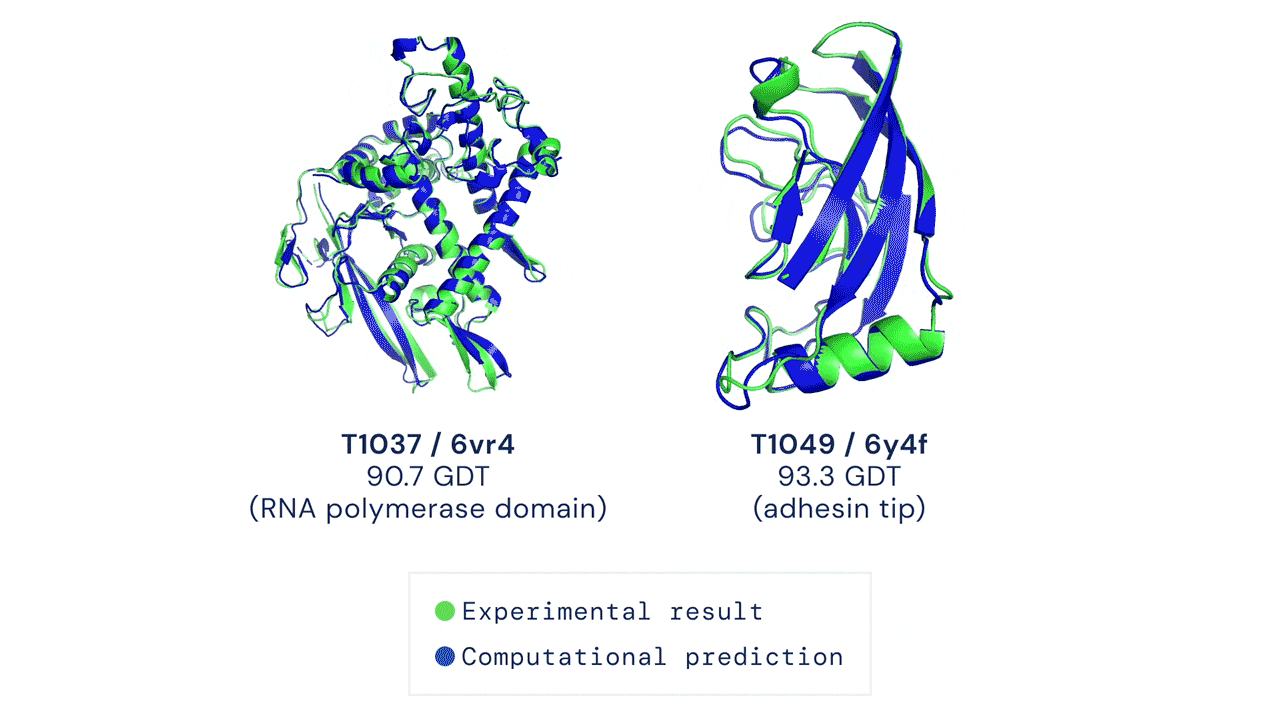
Two examples of protein targets in the free modelling category. AlphaFold predicts highly accurate structures measured against experimental result.
These exciting results open up the potential for biologists to use computational structure prediction as a core tool in scientific research. Our methods may prove especially helpful for important classes of proteins, such as membrane proteins, that are very difficult to crystallise and therefore challenging to experimentally determine.
This computational work represents a stunning advance on the protein-folding problem, a 50-year-old grand challenge in biology. It has occurred decades before many people in the field would have predicted. It will be exciting to see the many ways in which it will fundamentally change biological research.
We first entered CASP13 in 2018 with our initial version of AlphaFold, which achieved the highest accuracy among participants. Afterwards, we published a paper on our CASP13 methods in Nature with associated code, which has gone on to inspire other work and community-developed open source implementations. Now, new deep learning architectures we’ve developed have driven changes in our methods for CASP14, enabling us to achieve unparalleled levels of accuracy. These methods draw inspiration from the fields of biology, physics, and machine learning, as well as of course the work of many scientists in the protein-folding field over the past half-century.
A folded protein can be thought of as a “spatial graph”, where residues are the nodes and edges connect the residues in close proximity. This graph is important for understanding the physical interactions within proteins, as well as their evolutionary history. For the latest version of AlphaFold, used at CASP14, we created an attention-based neural network system, trained end-to-end, that attempts to interpret the structure of this graph, while reasoning over the implicit graph that it’s building. It uses evolutionarily related sequences, multiple sequence alignment (MSA), and a representation of amino acid residue pairs to refine this graph.
By iterating this process, the system develops strong predictions of the underlying physical structure of the protein and is able to determine highly-accurate structures in a matter of days. Additionally, AlphaFold can predict which parts of each predicted protein structure are reliable using an internal confidence measure.
We trained this system on publicly available data consisting of ~170,000 protein structures from the protein data bank together with large databases containing protein sequences of unknown structure. It uses approximately 16 TPUv3s (which is 128 TPUv3 cores or roughly equivalent to ~100-200 GPUs) run over a few weeks, a relatively modest amount of compute in the context of most large state-of-the-art models used in machine learning today. As with our CASP13 AlphaFold system, we are preparing a paper on our system to submit to a peer-reviewed journal in due course.

An overview of the main neural network model architecture. The model operates over evolutionarily related protein sequences as well as amino acid residue pairs, iteratively passing information between both representations to generate a structure.
When DeepMind started a decade ago, we hoped that one day AI breakthroughs would help serve as a platform to advance our understanding of fundamental scientific problems. Now, after 4 years of effort building AlphaFold, we’re starting to see that vision realised, with implications for areas like drug design and environmental sustainability.
Professor Andrei Lupas, Director of the Max Planck Institute for Developmental Biology and a CASP assessor, let us know that, “AlphaFold’s astonishingly accurate models have allowed us to solve a protein structure we were stuck on for close to a decade, relaunching our effort to understand how signals are transmitted across cell membranes.”
We’re optimistic about the impact AlphaFold can have on biological research and the wider world, and excited to collaborate with others to learn more about its potential in the years ahead. Alongside working on a peer-reviewed paper, we’re exploring how best to provide broader access to the system in a scalable way.
In the meantime, we’re also looking into how protein structure predictions could contribute to our understanding of specific diseases with a small number of specialist groups, for example by helping to identify proteins that have malfunctioned and to reason about how they interact. These insights could enable more precise work on drug development, complementing existing experimental methods to find promising treatments faster.
AlphaFold is a once in a generation advance, predicting protein structures with incredible speed and precision. This leap forward demonstrates how computational methods are poised to transform research in biology and hold much promise for accelerating the drug discovery process.
We’ve also seen signs that protein structure prediction could be useful in future pandemic response efforts, as one of many tools developed by the scientific community. Earlier this year, we predicted several protein structures of the SARS-CoV-2 virus, including ORF3a, whose structures were previously unknown. At CASP14, we predicted the structure of another coronavirus protein, ORF8. Impressively quick work by experimentalists has now confirmed the structures of both ORF3a and ORF8. Despite their challenging nature and having very few related sequences, we achieved a high degree of accuracy on both of our predictions when compared to their experimentally determined structures.
As well as accelerating understanding of known diseases, we’re excited about the potential for these techniques to explore the hundreds of millions of proteins we don’t currently have models for – a vast terrain of unknown biology. Since DNA specifies the amino acid sequences that comprise protein structures, the genomics revolution has made it possible to read protein sequences from the natural world at massive scale – with 180 million protein sequences and counting in the Universal Protein database (UniProt). In contrast, given the experimental work needed to go from sequence to structure, only around 170,000 protein structures are in the Protein Data Bank (PDB). Among the undetermined proteins may be some with new and exciting functions and – just as a telescope helps us see deeper into the unknown universe – techniques like AlphaFold may help us find them.
AlphaFold is one of our most significant advances to date but, as with all scientific research, there are still many questions to answer. Not every structure we predict will be perfect. There’s still much to learn, including how multiple proteins form complexes, how they interact with DNA, RNA, or small molecules, and how we can determine the precise location of all amino acid side chains. In collaboration with others, there’s also much to learn about how best to use these scientific discoveries in the development of new medicines, ways to manage the environment, and more.
For all of us working on computational and machine learning methods in science, systems like AlphaFold demonstrate the stunning potential for AI as a tool to aid fundamental discovery. Just as 50 years ago Anfinsen laid out a challenge far beyond science’s reach at the time, there are many aspects of our universe that remain unknown. The progress announced today gives us further confidence that AI will become one of humanity’s most useful tools in expanding the frontiers of scientific knowledge, and we’re looking forward to the many years of hard work and discovery ahead!
Notes
Until we’ve published a paper on this work, please cite:
High Accuracy Protein Structure Prediction Using Deep Learning
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Kathryn Tunyasuvunakool, Olaf Ronneberger, Russ Bates, Augustin Žídek, Alex Bridgland, Clemens Meyer, Simon A A Kohl, Anna Potapenko, Andrew J Ballard, Andrew Cowie, Bernardino Romera-Paredes, Stanislav Nikolov, Rishub Jain, Jonas Adler, Trevor Back, Stig Petersen, David Reiman, Martin Steinegger, Michalina Pacholska, David Silver, Oriol Vinyals, Andrew W Senior, Koray Kavukcuoglu, Pushmeet Kohli, Demis Hassabis.
In Fourteenth Critical Assessment of Techniques for Protein Structure Prediction (Abstract Book), 30 November - 4 December 2020. Retrieved from here.
We’re right at the beginning of exploring how best to enable other groups to use our structure predictions, alongside preparing a peer-reviewed paper for publication. While our team won’t be able to respond to every enquiry, if AlphaFold may be relevant to your work, please submit a few lines about it to alphafold@deepmind.com. We’ll be in contact if there’s scope for further exploration.



